Introduction
baytaAAR implements latent trait analysis within a
Bayesian Markov Chain Monte Carlo (MCMC) framework. It is intended to
estimate the age-of-death of adult individuals for whom one or several
ordinal traits related to the human aging process have been assessed. It
produces probability densities for the individual ages but also for the
respective population as a whole. baytaAAR has been
introduced and tested by Müller-Scheeßel et al. (2026), and there the basic idea of the model is
illustrated in the following figure:
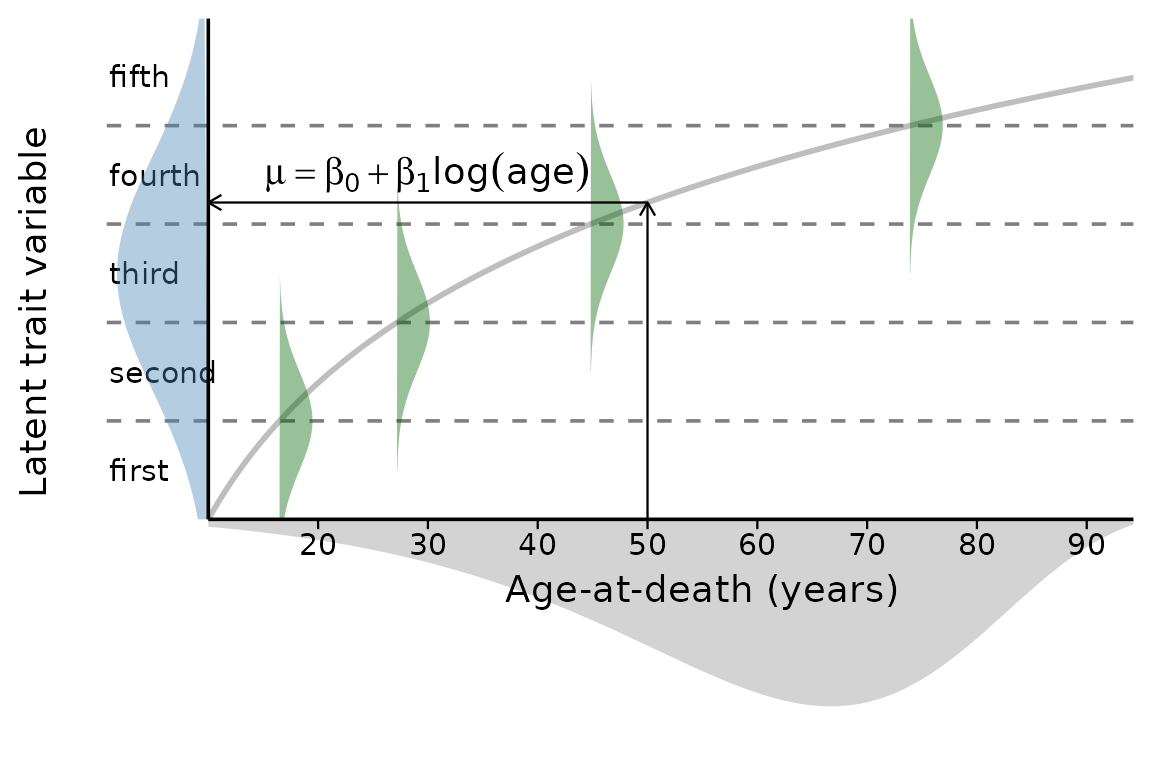
The figure schematically visualizes the ordered probit regression as a latent trait approach with age-at-death estimated from a Gompertz distribution (grey area). The green Gaussian distributions symbolize the transitions between the trait stages (here, arbitrarily, 5 stages, therefore 4 transitions). The regression line with its parameters intercept () and slope () determines the position of the latent trait variable. Please note that age is log-scaled, so the regression line appears curved. In practical applications, the latent trait variable does not need to be centered as it is here and the transitions are unlikely to be as evenly spaced as shown here.
More in-depth information on the background of and the rationale
behind baytaAAR can be found in the vignettes. One of them
discusses a real-life use-case with partly known ages-at-death.
Data layout
As input, the function bay.ta() assumes a
matrix() of trait expressions. In its simplest form, this
may contain only one column with a single trait as in the following
example (“auricular surface”):
data(sorsum_as, package = "baytaAAR")
head(sorsum_as)
#> id auricular_surface
#> 1 700 4
#> 4 704 5
#> 9 710 8
#> 10 711 4
#> 16 717 6
#> 17 719 3The example stems from the neolithic gallery grave Sorsum in Hessen/Germany. The auricular surface was the trait which was possible to assess most often (data from Moser et al. 2025, table S69), and it was assessed according to the method by Lovejoy et al (1985) which comprises eight levels. An overview of the distribution of the levels is given in the following plot:
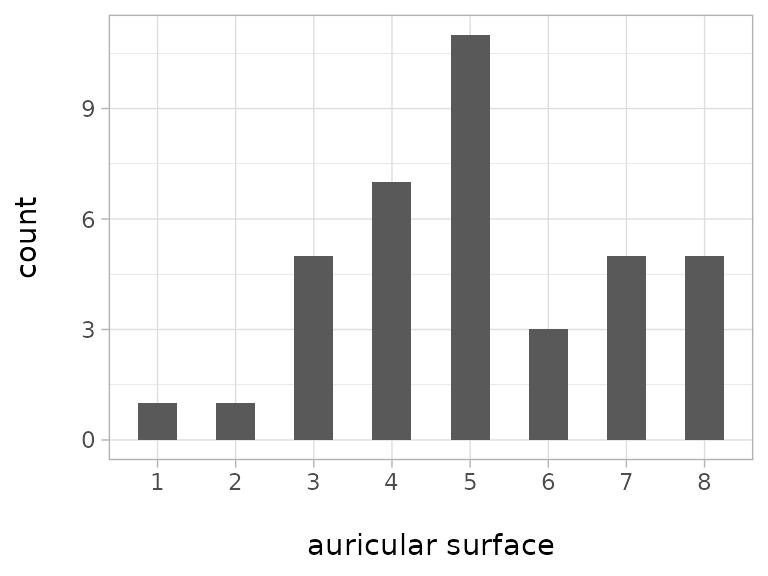
Level 5 is the most numerous while levels 1 and 2 are only present
once each. More on data input can be found in the
vignette("data_preparation").
Running a first analysis
We will now run a first analysis with bay.ta() with the
Sorsum data. We will leave most parameters at the default values and
only change the minimum age to 18 and the thinning interval
(thinsteps) to 1000. This number is multiplied
with the number of saved steps (1000) and the result
divided by the number of chains (default = 3) to obtain the total number
of iterations, minus the number of iterations used for burning-in. We
also set a seed for reproducibility.
sorsum_as_res <- bay.ta(
method = sorsum_as[,2],
minimum_age = 18,
thinSteps = 1000,
numSavedSteps = 1000,
seed = 1234
)
#> Starting Time: 16 Jun 2026 09:51:11
#> Defining model
#> Building model
#> Setting data and initial values
#> Running calculate on model
#> [Note] Any error reports that follow may simply reflect missing values in model variables.
#> Checking model sizes and dimensions
#> Checking model calculations
#> Compiling
#> [Note] This may take a minute.
#> [Note] Use 'showCompilerOutput = TRUE' to see C++ compilation details.
#> ===== Monitors =====
#> thin = 1: a, age.s, b, beta, beta0, thresh
#> ===== Samplers =====
#> RW sampler (85)
#> - age[] (38 elements)
#> - beta[] (1 element)
#> - beta0[] (1 element)
#> - b
#> - thresh[] (6 elements)
#> - ystar[] (38 elements)
#> Compiling
#> [Note] This may take a minute.
#> [Note] Use 'showCompilerOutput = TRUE' to see C++ compilation details.
#> running chain 1...
#> |-------------|-------------|-------------|-------------|
#> |-------------------------------------------------------|
#> running chain 2...
#> |-------------|-------------|-------------|-------------|
#> |-------------------------------------------------------|
#> running chain 3...
#> |-------------|-------------|-------------|-------------|
#> |-------------------------------------------------------|
#> Execution Time: 1.3 minutesThe analysis takes about 1 minute, depending on the computer power.
The console output informs about the monitored parameters
(a, age.s, b, beta,
beta0, thresh), the sampled nodes
(age, beta, beta0,
b, thresh and ystar), their
number and the used sampler which is RW for
Metropolis-Hastings sampling. The model output
(sorsum_as_res in this case) is a matrix with the number of
chains (three in the default case).
The output can be processed with any function able to deal with
coda::mcmc.lists() but baytaAAR provides some
functions for convenience to quickly achieve diagnostic results. One of
them is the function diagnostic.summary which does exactly
what its name suggests: a summary of diagnostic measures of the
parameters.
sorsum_as_res_diag <- diagnostic.summary(sorsum_as_res)
sorsum_as_res_diag |> head(10) |> knitr::kable(digits = 4)| PSRF Point est. | PSRF Upper C.I. | Mean | Median | Mode | ESS | MCSE | HDImass | HDIlow | HDIhigh | |
|---|---|---|---|---|---|---|---|---|---|---|
| M | 1.0235 | 1.0748 | 55.4075 | 60.1760 | 70.5845 | 620.9 | 0.7614 | 0.95 | 17.1554 | 81.7739 |
| a | 1.0226 | 1.0740 | 0.0091 | 0.0073 | 0.0029 | 621.7 | 0.0003 | 0.95 | 0.0002 | 0.0231 |
| age.s[1] | 1.0086 | 1.0309 | 45.1235 | 43.6152 | 40.7268 | 708.6 | 0.4788 | 0.95 | 20.8396 | 67.7356 |
| age.s[2] | 1.0041 | 1.0208 | 57.7845 | 57.2079 | 53.0281 | 666.3 | 0.5238 | 0.95 | 31.7521 | 81.9953 |
| age.s[3] | 1.0341 | 1.1155 | 82.9289 | 83.5910 | 82.9695 | 860.7 | 0.3295 | 0.95 | 65.2826 | 99.9182 |
| age.s[4] | 1.0123 | 1.0340 | 45.6070 | 44.4307 | 38.5401 | 647.7 | 0.5230 | 0.95 | 20.6297 | 70.8874 |
| age.s[5] | 1.0088 | 1.0322 | 67.1531 | 67.3303 | 72.0938 | 528.2 | 0.5269 | 0.95 | 44.4429 | 90.0163 |
| age.s[6] | 1.0083 | 1.0162 | 33.2717 | 30.7858 | 24.9060 | 627.2 | 0.4364 | 0.95 | 18.0831 | 55.3287 |
| age.s[7] | 1.0063 | 1.0238 | 73.2174 | 74.1248 | 74.9703 | 711.1 | 0.4262 | 0.95 | 52.1496 | 94.1982 |
| age.s[8] | 1.0015 | 1.0061 | 45.0487 | 43.3770 | 35.5461 | 671.8 | 0.4876 | 0.95 | 24.6571 | 70.3490 |
The diagnostic table gives a first impression of the result, above
the first ten rows are shown. diagnostics.max.min(),
another convenience function, displays minimum/maximum values of the two
quality measures Potential scale reduction factor (PSRF,
also called Gelman-Rubin statistic), a measure of chain mixing and the
Effective sample size (ESS), a measure of autocorrelation.
The PSRF value should be below 1.1, and the
ESS value larger than 10,000 [Kruschke (2015), 181; 184].
diagnostics.max.min(sorsum_as_res_diag)
#> PSRF_max PSRF_upper_max ESS_min
#> 1 1.070302 1.228565 33From the output of diagnostics.max.min() it is clear
that these values are not yet reached and that therefore more iterations
would be necessary.
Another measure to assess the quality of the simulations are
so-called ‘trace-plots’. They illustrate the mixing of the chains which
ideally should be nearly indistinguishable. For this, we use the
function bayesplot::mcmc_trace from the R package
bayesplot (Gabry et al.
2019). We also switch to the color scheme viridis
which makes distinguishing between chains easier.
bayesplot::color_scheme_set("viridis")
bayesplot::mcmc_trace(sorsum_as_res,
pars = c("age.s[1]", "beta[1]"), n_warmup = 300,
facet_args = list(nrow = 1, labeller = label_parsed))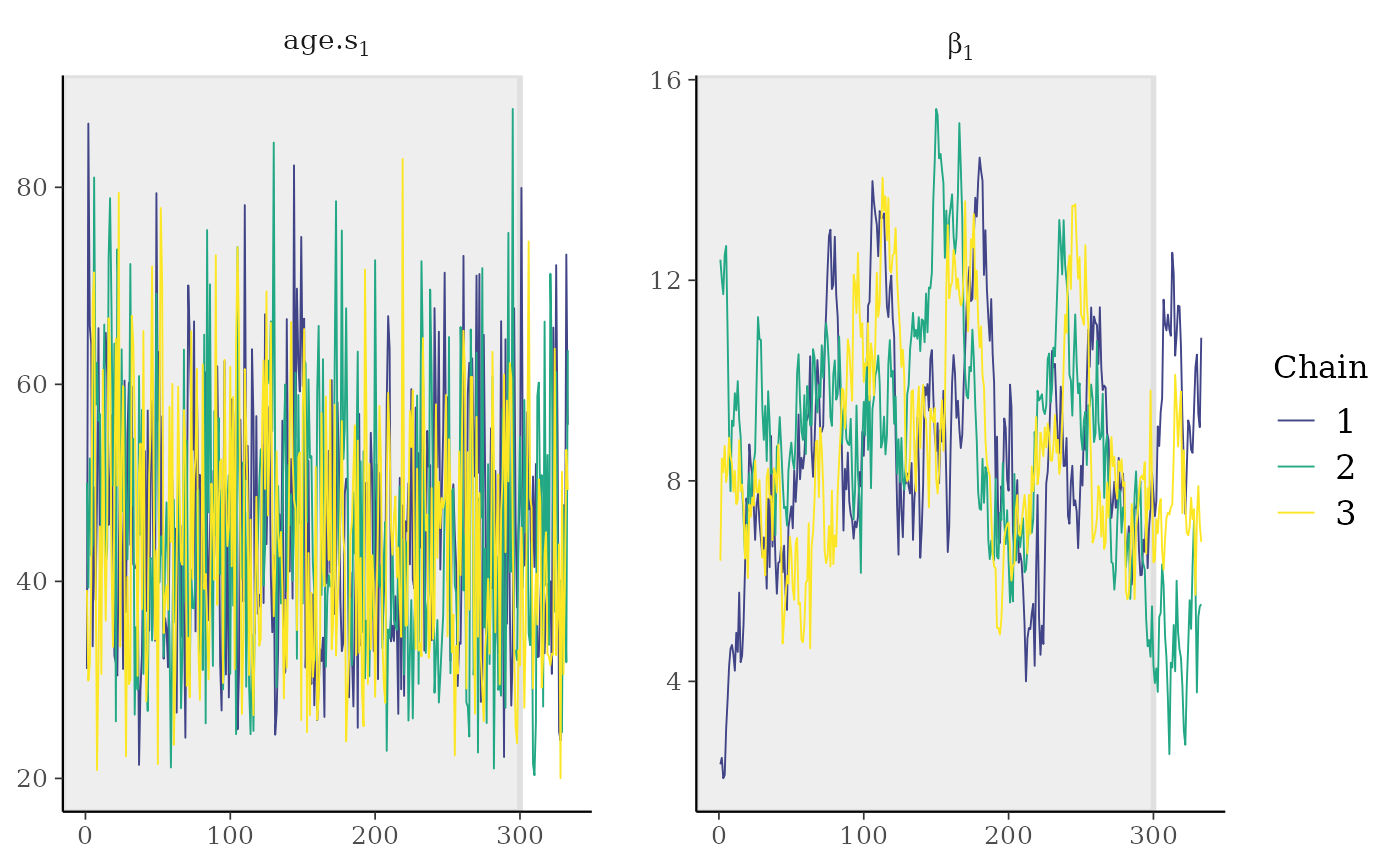
The left panel gives an impression how well-mixed chains should look
like for the first of the estimated ages. On contrast, the right panel
illustrates chains for the parameter beta, the slope of the
latent linear regression function, that have not yet well-mixed. Longer
chains are clearly necessary. This can also be illustrated with a plot
showing the Potential scale reduction factor (PSRF),
already mentioned above:
coda::gelman.plot(sorsum_as_res[, c("b", "beta[1]")])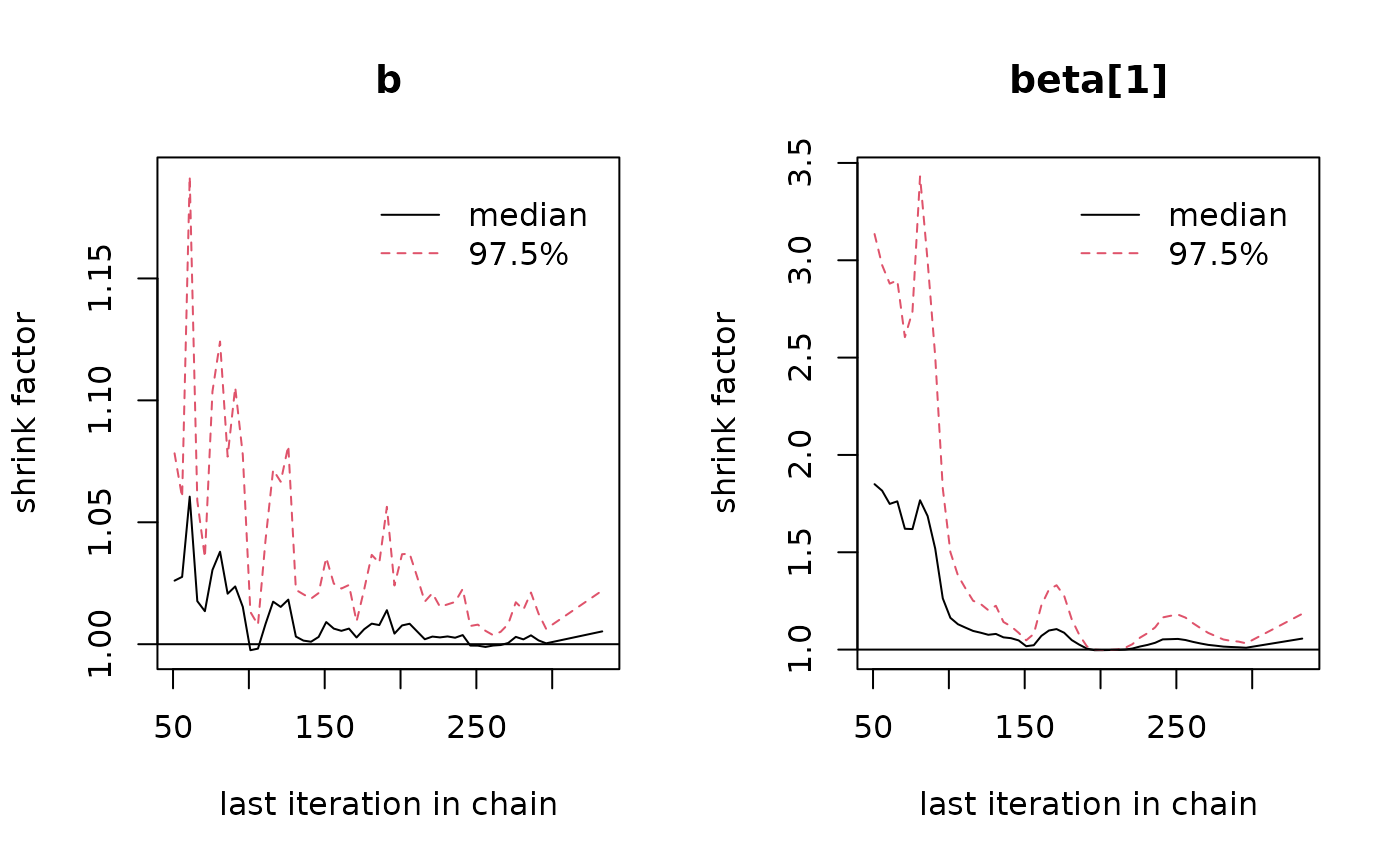
Ideally, the value of the PSRF should converge to 1, but
stay below 1.1. On the left panel for the parameter
b, this is clearly the case, but not so on the right panel,
again for the beta parameter (please note the difference in
scale of the y-axis!). This therefore also indicates that more
iterations are required.
From the minimum ESS value of around 25 (see above), it can be surmised that roughly 400 times more iterations are needed to get to a value of 10,000. This would increase runtime proportionally (approximately 200–250 minutes, equaling about 4 hours). However, because the resulting file would also be 400 times larger, some degree of thinning is necessary, so saving only, say, every 10,000th step (= thinning of 10,000). This in turn might increase autocorrelation which in turn will force you to run the model even longer.
In the current vignette, we forego this step at the moment. Instead,
we inspect the seven thresholds between the eight levels. For this, we
use the internal function threshold.chains() to compute the
threshold values for each iteration. The computation of the thresholds
on the age-scale is done outside of the MCMC simulation to reduce
computational cost and memory usage. The function returns a
coda::mcmc.list() which is further processed with first
diagnostic.summary() and then
threshold.matrix(). The latter is again a convenience
function to extract mean thresholds values from the output of
diagnostic.summary() which is particularly handy when
dealing with several traits.
thresholds <- threshold.chains(sorsum_as_res)
thresh_diag <- diagnostic.summary(thresholds)
threshold.matrix(thresh_diag) |> data.frame() |> knitr::kable(digits = 1)| X1 | X2 | X3 | X4 | X5 | X6 | X7 |
|---|---|---|---|---|---|---|
| 18 | 21.5 | 29.3 | 49.4 | 70.4 | 76.8 | 83.8 |
The thresholds of this trait (auricular surface) are unevenly spaced
within the age range of 18 and 100 years. The probability distribution
of the thresholds can conveniently visualised with e.g. the R-package
bayesplot. To avoid too glaring colors, we switch the color
scheme again:
bayesplot::color_scheme_set("gray")
bayesplot::mcmc_areas_ridges(thresholds, prob = 0.8, point_est = c("median"),
border_size = 0.2) +
theme_light() + xlim(18,100) + labs(x = "\nage-at-death (years)")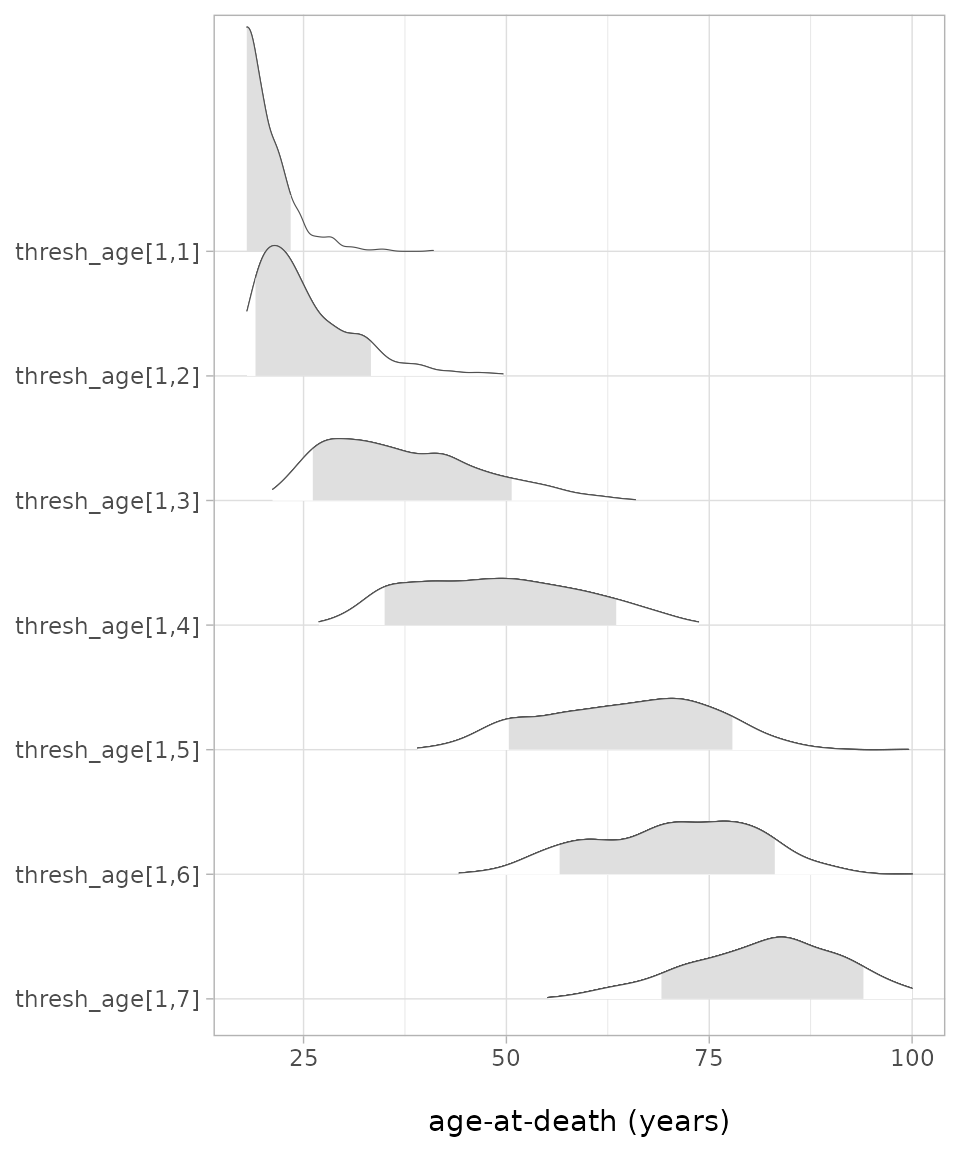 The resulting plot shows impressively the considerable overlap between
the thresholds, even when only the 80%-credible level is shown (grey
shaded areas), but also again that the thresholds are not evenly spaced.
Interesting would be a comparison with the thresholds of this trait
computed for other populations, keeping in mind that the current values
are not reliable as the quality measures were not yet met.
The resulting plot shows impressively the considerable overlap between
the thresholds, even when only the 80%-credible level is shown (grey
shaded areas), but also again that the thresholds are not evenly spaced.
Interesting would be a comparison with the thresholds of this trait
computed for other populations, keeping in mind that the current values
are not reliable as the quality measures were not yet met.
The function age.estim.summary() conveniently provides
summaries of age-related quantities like the mean estimated age, the
mean of the highest density interval of the estimated ages as well as
the parameters
,
,
and - derived from these two – the modal age M:
age.estim.summary(sorsum_as_res_diag) |> knitr::kable(digits = 3)| Mean | Median | Mode | 0.025 | 0.975 | |
|---|---|---|---|---|---|
| b | 0.044 | 0.041 | 0.029 | 0.020 | 0.075 |
| a | 0.009 | 0.007 | 0.003 | 0.000 | 0.023 |
| M | 55.407 | 60.176 | 70.585 | 17.155 | 81.774 |
| age_mean | 56.467 | 57.851 | 58.185 | 23.859 | 82.933 |
| hdi_diff | 41.918 | 45.582 | 46.995 | 17.680 | 50.300 |
The above table demonstrates that it makes a difference whether you
choose Mean, Median or Mode as
the measure of the mean.
To illustrate the distribution of some of the individual ages, this
time we rely on the functionality of the R package
tidybayes (Kay 2024). Its
function tidybayes::spread_draws() allows to subset the
chains in a single step to extract the estimated ages
(age.s). We limit here the age estimates to the first
seven.
sorsum_as_res |> tidybayes::spread_draws(age.s[age_number]) |>
subset(age_number < 8) |>
ggplot(aes(y = as.factor(age_number), x = age.s)) +
tidybayes::stat_halfeye(
.width = 0.95, point_interval = mode_hdi, fill = "lightgrey") +
scale_x_continuous(breaks = seq(10,100,10), limits = c(18, 100)) +
labs( x = "\nModal age-at-death (years)", y = "Individual no.\n" ) +
theme_light() +
theme(panel.grid.minor.x = element_blank(), text = element_text(size = 12))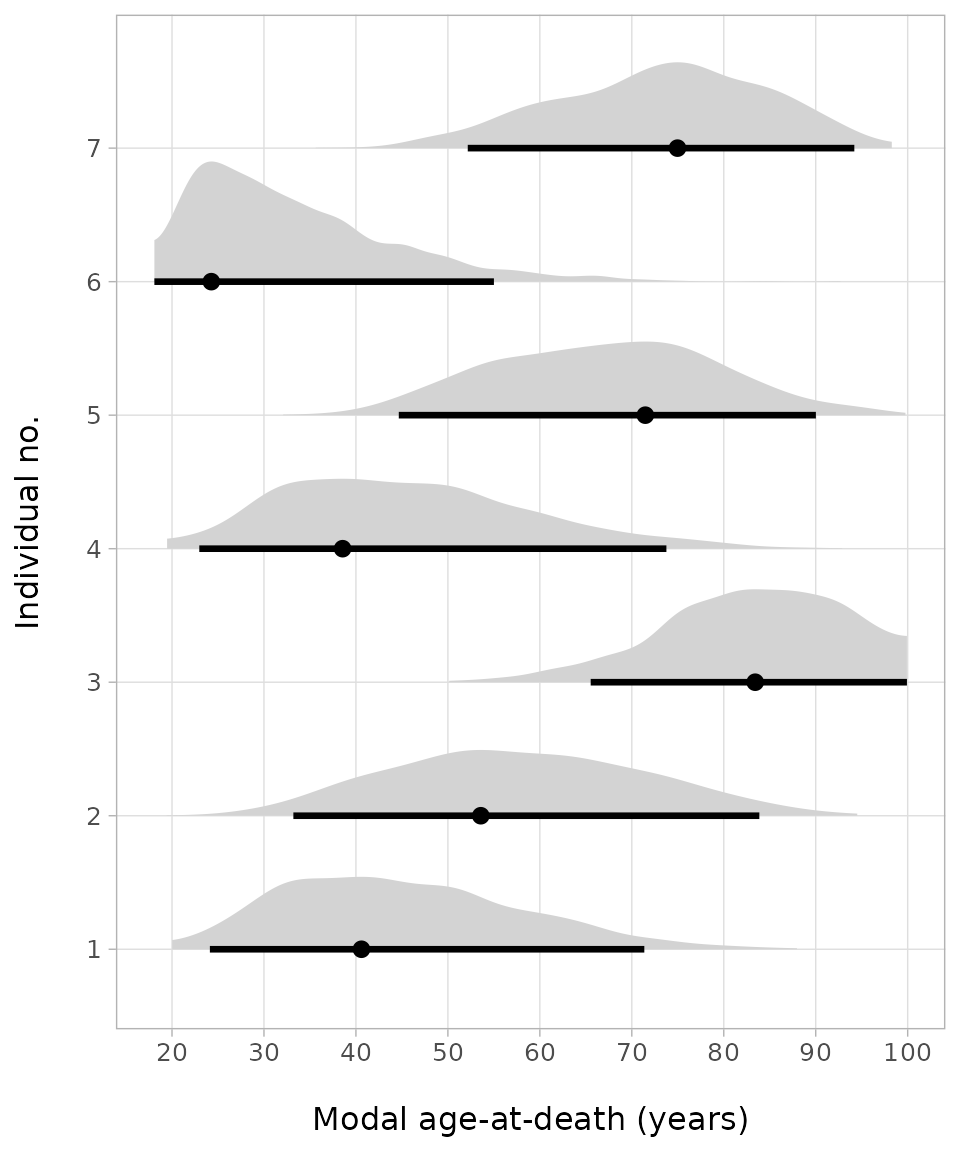
The plots of the first seven age estimates look similar to the threshold plots and allow immediate assessment of the spread of the probability distribution of the age estimates. So, for example, for the first individual with stage 4 of the auricular surface, the age mode is at about 40 years, but the 95%-credible range is between about 25 to 70 years.
Together with the individual age-at-death estimates,
bay.ta() also estimates the parameters of the underlying
Gompertz function,
and
:
ggplot() + ylab("density\n") +
geom_function(fun = function(x)
flexsurv::dgompertz(x - 18, sorsum_as_res_diag["b",3],
sorsum_as_res_diag["a",3])) +
xlab("\nAge in years") + theme_light() +
scale_x_continuous(breaks = seq(10,100,10), limits = c(18, 100)) +
theme(panel.grid.minor.x = element_blank(), text = element_text(size = 12))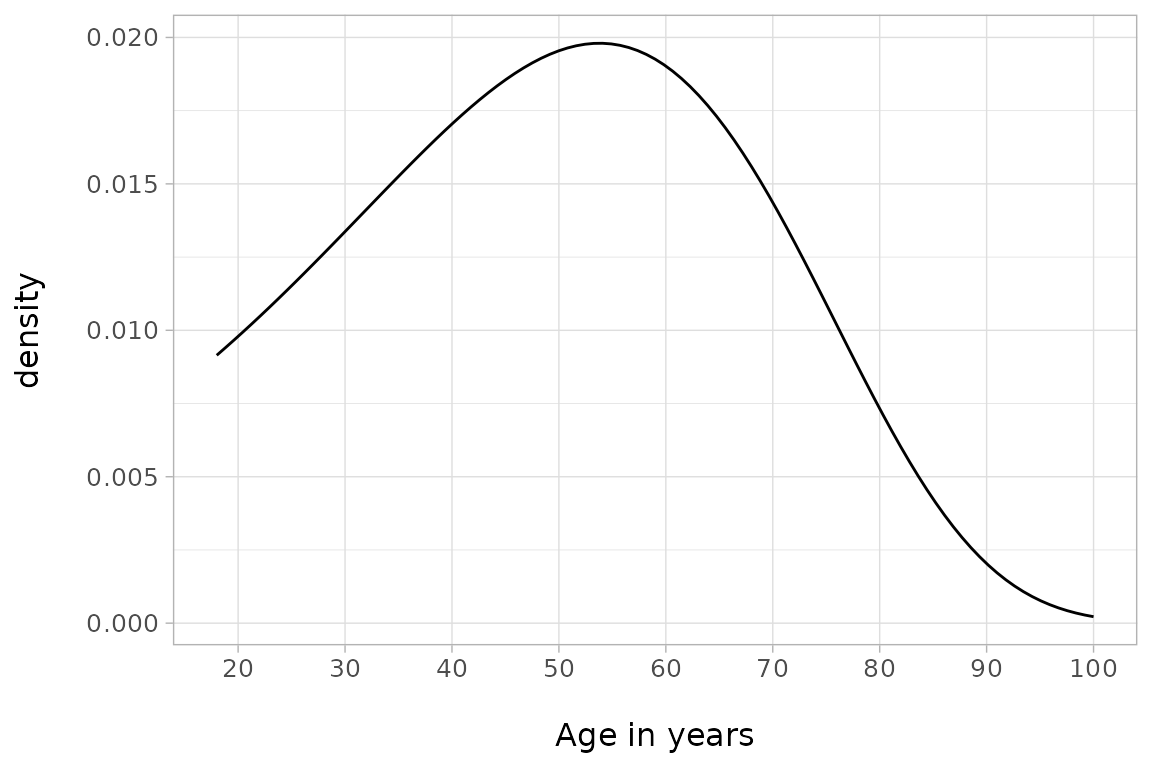
The Gompertz function provides a different perspective on the mortality structure of the population studied as it does not depend on individual point estimates of ages like, for example, Kaplan-Meier-diagrams. Please note that the maximum of the curve coincides with the arithmetic mean of the modal age M (55 years) in the table above.
Running the analysis with JAGS
Provided JAGS is
installed, bay.ta() can also be run with JAGS. For this, it
is sufficient to set the parameter framework to
JAGS. All other parameters remain unchanged.
sorsum_as_res <- bay.ta(
framework = "JAGS",
method = sorsum_as[,2],
minimum_age = 18,
thinSteps = 100,
numSavedSteps = 5000,
seed = 1234
)The JAGS model needs a little bit longer than the NIMBLE model but if you run the diagnostics you will see that the overall performance is superior. At this point, we leave the further analysis following the same steps as above to the reader.
Going further
More vignettes explain framework decisions
(vignette("computation_framework")), detail the model in
mathematical terms (vignette("mathematical_background")),
demonstrate how data sets with known age-at-death can be dealt with
(vignette("known_age")), provide a thoroughly worked
example (vignette("articles/worked_example")) and show how
the posterior probability densities can be grouped
(vignette("articles/groupings")).